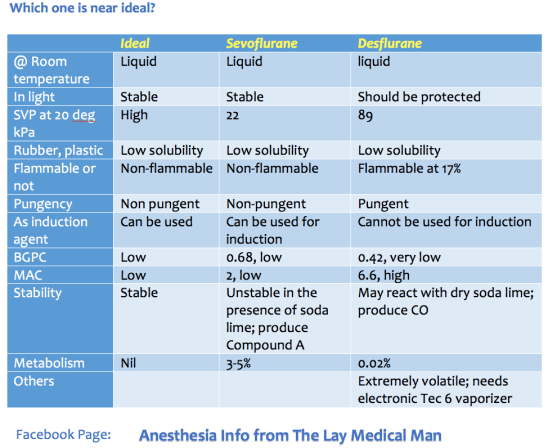
Drug administration in obese patients is difficult because recommended doses are based on pharmacokinetic data obtained from individuals with normal weights
With increasing obesity, fat mass accounts for an increasing amount of TBW, and the LBW/TBW ratio decreases
TBW is defined as the actual weight
IBW is what the patient should weigh with a normal ratio of lean to fat mass
IBW can be estimated from the formula: IBW (kg) = Height(cm) − x ( where x = 100 for adult males and 105 for adult females).
LBW is the patient’s weight , excluding fat
Male LBW = 1.1(weight)-128(weight/height)^2 (Weight in Kg and Height in cm)
Female LBW = 1.07 (weight) -148 (weight/height)^2
Regardless of total body weight, lean body weight rarely exceeds 100 kg in men and 70 kg in women
Below IBW, TBW and LBW are similar.
Adjusted body weight (ABW) Takes into account the fact that obese individuals have increased lean body mass and an increased volume of distribution for drugs.
It is calculated by adding 40% of the excess weight to the IBW : ABW (kg) = IBW (kg) + 0.4 [TBW (kg)]
Drugs with weak or moderate lipophilicity can be dosed on the basis of IBW or more accurately on LBW. These values are not same in obese; because 20–40% of an obese patient’s increase in TBW can be attributed to an increase in LBW. Adding 20% to the ‘estimated IBW based dose’ of hydrophilic medication is sufficient to include the extra lean mass. Non-depolarizing neuromuscular blocking drugs can be dosed in this manner.
In morbidly obese patients, the induction dose of propofol can be calculated on IBW.
In case of midazolam, prolonged sedation can occur from the larger initial dose needed to achieve adequate serum concentrations. #TheLayMedicalMan
Remifentanil dosing regimens should be based on IBW or LBW and not on TBW.
When using succinylcholine in obese adults or adolescents, dosage should be calculated on TBW
The antagonism time of neostigmine has been shown to be independent of TBW and BMI. Therefore, TBW can be used to calculate the dose.
Ref:Association of Anaesthetists of Great Britain and Ireland. Peri-operative management of the obese surgical patient 2015. Anaesthesia 2015, 70, pages 859–876.